




精选内容
-
胚胎染色体特纳氏综合征是在卵子精子一结合的时候就出现异常了吗?

 王玉玲医生的科普号2023年03月13日
王玉玲医生的科普号2023年03月13日 33
33
 0
0
 0
0
-
58.生长激素治疗特纳综合征有哪些好处?
一旦通过染色体检查确诊为特纳综合征后,可以用生长激素进行替代治疗,促进孩子身高增长直至达到理想身高;在达到青春期性发育启动年龄后,可以采用性激素进行替代治疗,促进第二性征的发育。使用生长激素治疗特纳综合征患者的目的或者说要达到的治疗目标是:尽早获得与年龄匹配的正常身高、重塑青春期生长加速、最终达到正常成年身高。目前已经有大量的研究证实早期注射重组人生长激素可以明显增加患儿的最终成人身高,干预得早的话甚至可以完全达到正常身高。在进入青春发育的年龄,可以联合使用生长激素和性激素,不仅可以进一步增加身高,而且可以逐步使患儿出现女性第二性征,并通过药物实现月经来潮。
王强医生的科普号 2022年11月19日239
0
1
2022年11月19日239
0
1
-
56.特纳综合征有哪些临床表现?
患者卵巢存在发育缺陷,导致成年后外阴幼稚,乳腺及乳头无发育,乳距增宽,无阴毛及腋毛生长,原发性闭经,无生育能力。大部分特的综合征患者身材矮小,母孕期及出生后生长明显缓慢,且没有青春期身高突增现象,未经治疗的患者终身高一般不超过143cm。患者面、颈、胸和背部皮肤常有黑痣,通贯手掌纹;表现为特殊面容,常有内眦赘皮和眼距过宽、塌鼻梁、有时耳轮突出、鲨鱼样口、腭弓高尖、小下颌,可伴有牙床发育不良;常见颈蹼、颈粗短和后发际低;部分存在言语障碍;部分患者表现为肘外翻、第4掌骨短、指趾弯曲、股骨和胫骨外生骨疣及指骨发育不良,偶尔可见膝外翻和脊柱侧弯现象;可合并心血管畸形以及泌尿系统畸形。
王强医生的科普号2022年11月19日231
0
1
-
性发育异常分类与诊断流程专家共识 中华预防医学会 生育力保护分会 生殖内分泌生育保护学组
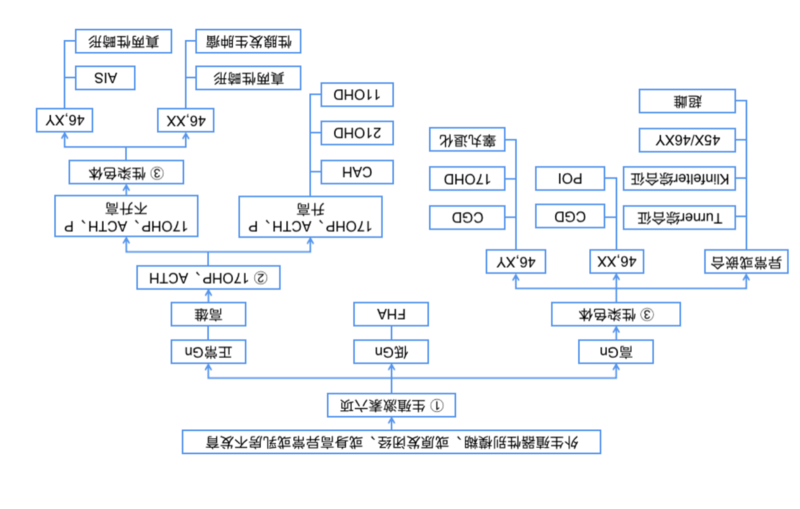
性发育异常(Disordersofsexdevelopment,DSD)是一种先天性异常,表现为性染色体、性腺或性激素性别的不一致[1]。因DSD就诊妇产科的患者,社会性别均为女性。DSD的发生率为新生儿的?1/4,500~5,000,其病因复杂,临床表现复杂多样,变化多端,对于不熟悉DSD的临床医生而言往往难以诊断和治疗[2]。人类的性别通常是由性染色体决定的,性染色体决定性腺性别,性腺的性质决定内外生殖器官的表型。但DSD患者可能存在不同形式的性染色体异常、性腺发育异常以及性激素合成和功能的障碍,形成三个层次的性别不匹配,即性腺性别与性染色体不匹配、内外生殖器官发育与性腺性别不匹配、以及性激素水平与性腺性别的不匹配。为了使广大临床医生,尤其是妇产科医生,能正确认识DSD、发现临床诊治线索、归类DSD,并最终确定DSD病因、提供恰当的治疗方案,特制定本共识,推荐适合于国内的DSD分类与诊断流程。一、DSD的分类长期以来,对此类疾病的诊断分类及命名一直难以统一,常以性腺的性质作为分类标准,将性染色体或性腺与表型不相符的称为“假两性畸形”。2005年在美国芝加哥国际会议上发布了DSD诊治共识,摒弃了既往“间性”、“两性人”的称谓,减少了歧视含义,也废除了“性反转”“XX男性”“XY女性”等词汇。进而将DSD按照染色体核型的不同分为性染色体核型异常DSD、46,XYDSD(主要与睾丸分化发育异常及雄激素合成、作用障碍有关)和46,XXDSD(主要与卵巢发育障碍、雄激素过多等有关)三大类[1](表1),这一命名与分类方法有利于在未明确诊断时给临床医师提供诊疗方向,是目前国际上认同度最高的分类法。此分类法大的分类较简单,但46,XYDSD和46,XXDSD两类中重复疾病种类较多,仅仅是染色体不同;另外其他类型,如46,XXDSD中MRKH综合征和泄殖腔发育异常等疾病属于生殖道发育异常,是否属于DSD尚有待商榷。表1.2005年芝加哥性发育异常诊治共识推荐的分类法[1,3-5](一)性染色体异常DSD45,X(特纳综合征和变异)47,XXY(Klinefelter综合征和变异)45,X/46,XY(混合性性腺发育不全,卵睾性DSD)46,XX/46,XY(嵌合体,卵睾性DSD)(二)46,XYDSD性腺(睾丸)发育异常:?(1)?完全型性腺发育不全(Swyer综合征)(2)?部分型性腺发育不良(3)?性腺退化(4)?卵睾性DSD雄激素合成或作用障碍:?(1)?雄激素生物合成缺陷a.?LH受体突变(LHCGR)(如Leydig细胞发育不全或无发育)b.?Smith-Lemli-Opitz综合征(DHCR7)c.?先天性磷脂增生(StAR突变)d.?胆固醇侧链裂解酶突变(CYP11A1)e.?3β羟基类固醇脱氢酶2缺乏(HSD3B2)f.?17羟基类固醇脱氢酶缺乏(HSD17B3)g.?5α还原酶2缺乏(SRD5A2)(2)?雄激素作用缺陷(AR)a.??完全型雄激素不敏感综合征(CAIS)b.??部分型雄激素不敏感综合征(PAIS)其他类别:?(1)?综合征相关的男性生殖道发育异常(如泄殖腔异常,Robinow综合征,Aarskog综合征,手-足-生殖器综合征,腘翼状赘肉综合征)(2)?持续性苗勒管综合征(AMH,AMHR2)(3)?睾丸退化综合征(4)?与激素缺陷无关的(孤立的)尿道下裂(CX或f6);(5)?先天性低促性腺激素性性腺功能减退(6)?隐睾症(INSL3,GREAT)(7)?环境影响(三)46,XXDSD性腺(卵巢)发育障碍:(1)?卵睾性DSD;(2)?睾丸性DSD(如SRY+,重复SOX9,RSP01)(3)?性腺发育不全雄激素过多:?(1)?胎儿肾上腺a.?21-羟化酶缺乏(CYP21A2)b.?11β-羟化酶缺乏(CYP11B1)c.?3β-羟基类固醇脱氢酶2缺乏(HSD3B2)d.?细胞色素P450氧化还原酶缺乏(POR)e.?糖皮质激素受体突变(2)?胎儿胎盘a.?芳香化酶缺乏(CYP19)b.?细胞色素P450氧化还原酶缺乏(POR);(3)?母体a.?男性化肿瘤(如黄体瘤)b.?外源性雄激素药物其他类别(1)?综合征相关(如泄殖腔异常)(2)?阴道闭锁(MRKH综合征);(3)?MURCS(苗勒管,肾,颈胸部躯体异常),其他综合征(4)?子宫异常(如MODY5)(5)?阴唇粘连?美高梅注册/北京协和医院葛秦生教授总结多年的临床经验与基础研究,于1994年提出根据性发育过程中三个最关键的环节:性染色体、性腺和性激素作为分类的基础,将此类复杂疾病按病因分为三大类的方法[6](表2),相比之下,我们认为葛氏分类法更简单、更实用、更方便,逻辑性也更强,推荐国内同行使用。葛氏分类法第一次彻底地抛弃了“假两性畸形”的混乱概念,根据疾病病因进行分类,并可据此进行相关病因的基础研究,避免了资源浪费,相比芝加哥共识,这一观念提前了10多年。1975年~2018年间,美高梅注册/北京协和医院妇科内分泌组共收集了临床所见各种DSD病例近千例,按此分类法均可适当地进行分类,在实际应用中证明了此分类法条理清晰,简单明了,易于掌握,便于正确诊断和处理。葛氏分类法是一开放的分类法,虽未包括所有罕见类型,但亦不外乎这三个层次。表2?美高梅注册/北京协和医院葛秦生性发育异常分类(一)性染色体异常:包括性染色体数与结构异常1.?特纳综合征2.XO/XY性腺发育不全3.超雌(47,XXX等)4.真两性畸形(嵌合型性染色体)5.曲细精管发育不良(Klinefelter)综合征(二)性腺发育异常1.XX单纯性腺发育不全2.XY单纯性腺发育不全(完全型与部分型)3.真两性畸形(46,XX或46,XY)4.睾丸退化(三)性激素量与功能异常1.雄激素过多1)?先天性肾上腺皮质增生a.?21-羟化酶缺乏(CYP21A2)b.?11β-羟化酶缺乏(CYP11B1)c.?3β-羟基类固醇脱氢酶2缺乏(HSD3B2)d.?细胞色素P450氧化还原酶缺乏(POR)2)?芳香化酶缺乏(CYP19)?3)?早孕期外源性雄激素过多4)?孕母男性化肿瘤(如黄体瘤)2.雄激素缺乏(合成酶缺乏)1)?17α-羟化酶缺乏(完全型与部分型)2)?5α-还原酶缺乏3)?Leydig细胞发育不全4)?类脂性先天性肾上腺皮质增生症(StAR突变)5)?胆固醇侧链裂解酶缺乏(CYP11A1)3.?雄激素功能异常(雄激素不敏感综合征)1)?完全型2)?部分型二、DSD的诊断流程临床上遇到下列情况:原发闭经、身高过矮或过高、第二性征不发育、外生殖器性别不清和特殊的躯体特征等,要考虑DSD存在的可能,需通过以下流程,进行诊断与鉴别诊断。(一)病史询问围绕患者的主诉,仔细了解现病史、孕期用药史及家族史,特别是询问外生殖器、乳房、身高的变化历史,以及有无高血压病史。外生殖器出生后就发现异常,以后未再进展,常见于胚胎期的雄激素异常,如睾丸退化、孕早期使用大剂量雄激素等;出生后发现异常,以后又加重,提示出生后性腺仍有功能,常见于21-羟化酶缺乏导致的先天性肾上腺皮质增生(Congenital?adrenalhyperplasia,CAH)、部分型雄激素不敏感综合征(Androgeninsensitivitysyndrome,AIS)、真两性畸形(或卵巢-睾丸型DSD)等。青春期后有自动乳房发育,提示有内源性雌激素作用;如是用药后发育或乳房整形,病因等同于无发育;乳房无发育,提示缺乏内源性雌激素作用,常见于性腺发育不全;乳房有发育、但乳头发育不良,常见于AIS。身高从小一直偏矮见于Turner综合征;较同龄人先高后矮、合并有男性化表现的性早熟常提示21-羟化酶缺乏CAH。身高明显超过同龄人者合并乳房不发育,提示缺乏雌激素作用,常见于性腺发育不全或超雌。孕早期服用含有雄激素作用的药物或母体分泌雄激素肿瘤,可导致出生后外生殖器异常,如阴蒂长大、后联合融合,但出生以后不会再进展发育。有些DSD种类有遗传家族史,如CAH为常染色体隐性遗传病,而AIS为X-连锁隐性遗传病,常见于母系家族。(二)体格检查要注意观察身高、肤色、喉结、嗓音,以及全身的特殊体征,并注意外阴阴蒂大小、后联合高低、性腺部位和盆腔检查。检查有无血压升高,CAH中的11-羟化酶缺乏、17-羟化酶缺乏均可有严重的高血压,但患者及家属可能从未发现或重视。如身高低于150cm,高度怀疑特纳综合征或常见的21-羟化酶缺乏CAH可能;而身高过高提示患者有男性XY染色体或多个X染色体(如超雌)存在的可能。特纳综合征患者常伴有面部多痣、颈蹼、肘外翻等特殊体征。而21-羟化酶缺乏CAH常有肤色深、嗓音低沉、新出现喉结、阴蒂肥大等。注意腋毛、阴毛的发育,没有阴腋毛提示缺乏雄激素作用;阴腋毛过多则提示雄激素过高或作用过强。阴蒂增大、后联合融合提示雄激素作用过强、较久。检查腹股沟或大阴唇内是否可触及包块,如是性腺则为睾丸或卵睾,卵巢不会下降到此。注意检查有无阴道、盲端阴道、有无宫颈、子宫。(三)辅助检查1.?生殖激素测定DSD患者通常都需要检测生殖激素六项,包括黄体生成素(LH)、卵泡刺激素(FSH)、泌乳素(PRL)、雌二醇(E2)、孕酮(P)、睾酮(T)。首先可以根据促性腺激素(LH、FSH)的水平区分出促性腺激素升高、正常以及降低三种类型。常见的DSD以促性腺激素升高和促性腺激素水平正常为主。雄激素是影响外生殖器分化的主要激素,雄激素主要包括T、雄烯二酮(A)、硫酸脱氢表雄酮(DHEA-S)、双氢睾酮(DHT)等。孕酮水平持续升高(任何时间点测定P均超过排卵后的水平?3ng/ml),排除常见的怀孕、妊娠产物残留、排卵等病因后被称为高孕激素血症,则是怀疑CAH的重要线索,可通过检测促肾上腺皮质激素(ACTH)、17-羟孕酮(17OHP)加以验证。涉及CAH相关的其他代谢酶缺乏,可通过质谱法检测的类固醇激素谱对比肾上腺类固醇激素合成途径,推断酶缺乏的部位。对于青春期前的患者,人绒毛膜促性腺激素(hCG)和尿促性腺激素(hMG)刺激试验对于评估性腺的功能很有必要。鉴别肾上腺来源的高雄激素血症,有时会采用地塞米松抑制试验进行鉴别诊断。2.?血清电解质检查包括K+、Na+、Cl-测定。CAH患者中,失盐型21-羟化酶缺乏可有高血K+、伴低Na+、低Cl-;而17-或11-羟化酶缺乏患者可有高血Na+、低血K+。3.?影像学检查最常用的是盆腔超声检查,未婚的经直肠超声较经腹超声好,了解子宫及性腺的结构和位置,必要时腹股沟区B超,及肾脏及肾上腺B超。但受限于子宫较小以及医生的经验不足,可能存在误报(超声报告有子宫而实际无子宫、报卵巢组织实际可能是睾丸组织等)和漏报(超声报告未见子宫而实际上有小子宫、未见到性腺组织等)的可能,还需结合其他临床信息综合判断。MRI?对DSD患者肾上腺增生与生殖器官的特异性及灵敏度均较高,其中子宫在93%左右,阴道在95%,阴茎在100%,睾丸在88%,卵巢在74%[7],但对于排除腹腔内性腺仍然不是绝对可靠[8]。有无子宫取决于胚胎发育早期有无睾丸分泌的抗苗勒管激素(Anti-mullerian?hormone,?AMH),对于缺乏AMH的DSD个体,绝大多数是会有子宫的,即使检查时偏小,多数是缺乏性激素所致,具有后天发育的潜能,所以对疾病的病因确认才能保证不错误切除患者有生育潜能的器官,而且不漏掉可能恶变的器官,这也是DSD疾病明确病因和正确分类的重要性所在。4.?染色体核型及基因检测染色体核型的检测对于DSD的诊断至关重要,临床常用的方法是染色体G带显色法,不仅可以检测染色体数目上的变化,而且可以观察到缺失、重复、倒位、异位等结构上的异常。染色体的检测结果不仅与诊断归类密切相关,更重要的是识别含Y染色体成分的患者,按女性生活需要进行性腺切除以预防肿瘤的发生[9],关乎治疗决策和预后。值得注意的是,少数病例的核型检测虽然没有Y染色体,但存在标记染色体成分,需要进一步行SRY基因的检测,如SRY(sex-determiningregionofY-chromosome,Y染色体性别决定区)基因阳性,应按含Y染色体成分处理[10]。随着基因检测技术的快速发展和对DSD作为公共卫生问题的认识提高,在过去10年中对DSD分子水平的病因认识有了长足的进步。对DSD患者进行相关病因基因的检测,为确定病因、鉴别诊断、改善预后和遗传咨询提供了条件[11-13]。但由于DSD的发病机制复杂,目前的基因检测技术并不能全部发现其基因突变,故其在临床的应用与价值尚需进一步的探索与研究,应在专业人士的指导下,结合临床诊断、有目的地开展与应用。5.手术探查对诊断不清楚、或怀疑真两性畸形诊断、有手术探查指征的DSD患者,手术探查对明确诊断、去除病灶、评估生殖预后有重要的意义[14]。真两性畸形的确诊,除个体有男女两性的临床表现外,还必须术中在一个个体中同时发现有睾丸和卵巢组织的存在、并且在活检或组织切除的病理切片中能同时见到卵泡和曲细精管。【共识要点】??DSD的诊断从症状、体征入手,结合影像学、激素水平和染色体、基因等辅助检查手段,绝大多数都可明确病因诊断、正确归类(见图)。?Gn:促性腺激素;FHA:功能性下丘脑性闭经;CGD:完全型性腺发育不全;POI:早发性卵巢功能不足;17OHP:17-羟孕酮;P:孕酮;ACTH:促皮质激素;CAH:先天性肾上腺皮质增生;21OHD:21-羟化酶缺乏;11OHD:11-羟化酶缺乏;AIS:雄激素不敏感综合征;46,XX、46XY:染色体结果。??对于含有Y染色体或Y染色体成分的DSD患者,按女性生活的切除性腺是必要和重要的处理,理论上有子宫的尽量保留子宫,经规范的治疗后可有月经,且通过辅助生育技术可实现生育。??美高梅注册/北京协和医院葛氏分类法,经过多年临床验证,简洁实用,适于推广,同时也具有开放性,新发现或罕见的病种也可归入不同层次的类别中。参考文献省略
 田秦杰医生的科普号
田秦杰医生的科普号 2022年10月13日1378
0
1
2022年10月13日1378
0
1
-
我是特纳综合症,在吃芬吗通的时候前两年月经正常,但是后面经期好久才干净,不知道是怎么回事呀
 妇幼营养与骨健康大讲堂
妇幼营养与骨健康大讲堂 2022年07月07日227
0
0
2022年07月07日227
0
0
-
特纳综合征中国专家共识(2022年版)
特纳综合征中国专家共识(2022年版)为进一步规范和提高临床医师对TS的诊治水平,中国人体健康科技促进会生育力保护与保存专业委员会组织专家进行了专题讨论,达成以下共识,以供临床参考。什么是特纳综合征?特纳综合征(TS)是一种女性表型的先天性染色体异常疾病,其中一条X染色体完整,另一条性染色体完全或者部分缺失,其临床表型多样。TS的临床表现TS是一种一条X染色体正常,另一条性染色体完全或部分缺失的先天性染色体异常疾病,临床表现主要为身材矮小,原发性性腺发育不良及其他先天畸形如头面部特殊面容、肘外翻等,并同时合并其他系统疾病。(A)身材矮小95%~100%的TS患者表现为身材矮小,通常在胎儿时期即出现宫内轻度生长受限;在婴儿期和整个儿童期,身高增长依然缓慢,且明显低于同年龄正常人群平均身高2个标准差(-2SD);无青春期身高突增;TS患者的成年身高与父母身高相关,但明显低于正常女性。原发性性腺发育不良TS患者常伴有原发性性腺发育不良,表现为女性幼稚外阴、生殖器和第二性征不发育。15%~30%的TS患者最初有乳房发育,然而在青春期停滞;或者在青春期发育后,出现继发性第二性征发育迟缓,进而发生卵巢早衰。发育异常及先天畸形TS患者常具有特殊的躯体特征,如颈蹼、盾状胸、肘外翻,皮肤黑痣,内眦赘皮,上眼睑下垂,下颌小,部分患者有第4掌骨短、指趾弯曲、股骨和胫骨外生骨疣及指骨发育不良等。自身免疫性疾病TS患者更易发生自身免疫性疾病,如自身免疫性甲状腺炎、糖尿病、幼年特发性关节炎、炎症性肠病、乳糜泻等,且发病风险随年龄增长而增加。智力及神经认知功能大部分TS患者智力正常,智力障碍的发生可能与小的环状X染色体相关。部分TS患者可能伴有特殊类型的学习障碍,包括注意缺陷、多动障碍、学习障碍以及特异性的神经心理缺陷,如视觉-空间组织缺陷、社会认知障碍、执行功能障碍、运动缺陷等。TS的诊断外周血染色体核型分析是诊断TS的金标准,此外需结合生长缓慢的病史、第二性征发育不良的表型、性激素水平以及影像超声检查等辅助结果综合诊断。(A)出生前诊断★染色体核型检测所有疑似TS的患者均应接受染色体核型检测以明确TS的诊断。外周血染色体核型分析是诊断TS的金标准染色体核型分析筛查适应证见表1。★病史应着重询问患者母亲孕期超声检查有无水肿胎及胎儿颈部囊性淋巴瘤、浆膜腔积液、颈项透明层增厚等异常病史,有无身材矮小及青春期延迟等临床表现。★体格检查测量血压、视力、听力、身高、体重指数及乳腺、外生殖器Tanner分期等,评估有无TS相关的发育异常或先天畸形。★辅助检查主要进行肝肾功能、血糖等一般检查,还应进行性激素、甲状腺功能、心血管、泌尿系统、消化系统、骨密度检查,全面了解患者的基本情况。产前诊断★超声检查超声和母体血清筛查只能辅助诊断TS,最终仍需要染色体核型分析确诊。★遗传学检查羊水细胞或外周血淋巴细胞染色体核型分析是诊断的金标准。约半数TS为X单体型(45,X0),20%~30%为嵌合型(45,X0/46,XX),其余多为另一条X染色体结构异常。★无创产前诊断当怀疑TS时,应进一步筛查母体染色体核型、羊膜穿刺术、胎儿超声心动图和遗传咨询。★植入前基因筛查植入前基因筛查可以在胚胎植入前对胚胎进行染色体异常的筛选,因此能提高体外受精的成功率。TS的治疗促生长治疗促生长治疗的目标是使患者达到遗传靶身高;使其成人身高适度增加。rhGH治疗建议从4~6岁开始,推荐剂量0.35~0.42mg(/kg·周),不宜超过0.42mg/(kg·周),当达到满意身高、骨龄>13岁或年生长速率<2cm/年,建议终止rhGH治疗(表2);当治疗效果欠佳时,可考虑联合用药,推荐司坦唑醇或氧雄龙。治疗期间需严密监测血糖、胰岛素、甲状腺功能、IGF-1水平。(A)性激素补充治疗推荐性激素补充治疗从TS患者骨龄11~12岁开始(表3),雌激素剂量以成人生理剂量1/8开始,2~3年内逐步增加至成人生理剂量。当出现突破性出血时,后半周期添加孕激素10~14d,优先推荐天然或接近天然的孕激素,如地屈孕酮或微粒化黄体酮。(A)TS的生育力保护及生育策略目前TS患者的生育策略主要包括:(1)卵巢组织冷冻保存。(2)自然妊娠。(3)赠卵辅助生殖技术。目前赠卵仍是有生育要求的TS患者妊娠的主要途径之一。卵巢组织冻存是一种新的被国际、国内指南推荐的TS儿童及青春期前女孩唯一的一种生育力与卵巢功能保护方法。(A)TS的围妊娠期管理TS患者生育前必须充分接受产前诊断、心血管科、内分泌科、母胎医学、生殖中心等多学科专家团队综合评估及围妊娠期管理。(A)TS的中医治疗中医治疗在明确TS诊断后,根据患者的体质、年龄、症状与舌脉,辨证论治,治疗以补肾养肝益脾为主。(B)TS的并发症TS患者的并发症治疗需关注多器官、多系统综合诊治,建议有条件的医疗机构建立多学科诊疗(MDT)门诊,并进行个体化监测及长期随访(A)。TS的随访TS的临床表型复杂多样,可累及多系统、多器官,部分并发症的发生率如主动脉扩张、高血压、糖尿病等随年龄增长而增加。因此,对TS的临床监测随访宜贯穿TS诊治的整个过程。主要的基线评估和监测频率见表4。TS多学科诊疗模式的建立TS患者的合并症在儿童或青少年期可能尚未出现,然而随着年龄增长,不仅合并症发生率增高,还存在其他社会心理等问题。因此,我们建议有条件的医疗中心进行多学科一站式综合诊治。这些学科应包括内分泌科、妇科、心血管科、遗传及产前诊断科、心理咨询科、耳鼻喉科、皮肤病科和消化内科等。同时应对TS患者的生活方式(如运动、饮食和体重控制等)、社会心理和生育计划等进行健康宣教及专业评估,制定个性化诊疗方案。来源:中国实用妇科与产科杂志,2022,38(4):424-433.
 任卫东医生的科普号
任卫东医生的科普号 2022年05月15日2158
1
5
2022年05月15日2158
1
5
-
夫妇正常为什么胎儿染色体有问题?
夫妇及家庭以上几代的染色体都正常,胎儿会发生染色体疾病吗? 胎儿期常见的染色体疾病包括唐氏综合征(21三体综合症)、特纳综合征(45,X综合症)、18三体综合征、13三体综合征、16三体综合症、22三体综合症等。 上述综合征的发生绝大多数与孕妇年龄有关,只有极少部分从上代遗传而来,因此夫妇及家庭以上几代的染色体都正常,不能保证胎儿不会生染色体疾病。 随着孕妇年龄增大,卵细胞在成熟过程中染色体配对和分离会出现差错,带有多余染色体的卵细胞与正常精子受精后将形成染色体不正常的胎儿。
 翟志瑾医生的科普号
翟志瑾医生的科普号 2021年12月01日620
0
2
2021年12月01日620
0
2
-
它,不仅会导致孩子身材矮小,还影响未来生育!
前几天看了一本书,名叫《登梯之旅:我与特纳综合征》(Ladders to Climb: My Journey with Turner Syndrome),讲述的是一位特纳综合征患者,从出生到成长的故事。作者在书中写道:“我在学校被同学取笑、岐视,还会遭受各种各样异样的眼光。但是我一定要更坚强、更勤奋,我要像一个正常的女孩一样,快快乐乐地生活。” 我不禁想起这些年我在门诊遇到的几十个特纳综合征患儿,每一个患儿就诊、治疗、成长的背后,都是一个家庭的心酸曲折的故事。 2008年冬天,某天上午我接诊了一位来自毕节的女性患儿萱萱。患者7岁3个月,身高103厘米
付晓玲医生的科普号 2021年04月12日1299
0
0
2021年04月12日1299
0
0
-
“小”女孩的烦恼-Turner's综合征
小文今年14岁,但身高只有140cm,远比同龄人矮小。而且,当同龄女孩都已长成亭亭玉立的少女时,小文外表仍象个小女孩,胸部平平,月经也未来潮。妈妈带小文去妇科检查,化验结果出来后,医生告诉她们,小文患的是一种染色体疾病,称为Turner’s(特纳氏)综合征。上海市第一妇婴保健院妇科王哲蔚Turner’s综合征是一种因染色体异常引起的综合征,主要表现为先天性性腺发育不全,在人群中的发生率为1/2500-10000。正常女性的染色体核型是46XX(即性染色体由2条X染色体组成),但这类患者的染色体核型为45X(缺1条X染色体)。患者主要表现有(1)身材矮小,如不治疗,一般成年身高不会超过150cm(2)不同程度的性发育幼稚,缺乏第二性征,蹼状颈,肘外翻。(2)可伴有心血管或肾脏畸形,或自身免疫性疾病。(3)先天性卵巢功能衰竭。(4)智力正常,但可能有特殊领域的认知障碍。基因型为纯45X型的患者(占50%左右)症状较重,仅近20%出现性发育和月经。基因型为嵌合型的(45X/46XX,45X/47XXX,45X/46XXp-或46XXq-)症状较轻,有70%出现性发育,2%有生育力。小文妈妈非常困惑:这孩子出生时虽然体重偏轻,但和其他孩子一样健康活泼。这个诊断对她们来说简直是晴天霹雳,的确,大多数Turner’s综合征的宝宝出生时外观和正常的女婴没什么区别,在儿童期除了生长缓慢,往往无其他异常。很难早期发现。那家长们如何尽早发现呢?1、孕期:一旦孕期B超发现胎儿水肿,,颈部透明度增加,囊状水瘤,主动脉缩窄,左心室畸形,肾脏畸形,短头畸形,羊水过少等,可进行羊水染色体检查排除包括Turner’s综合征在内的染色体疾病,但即使孕期已确诊,出生后的婴儿仍要接受血染色体检查(出生后的染色体核型检验报告可能与孕期不一致,以出生后的为准)。孕期未发现异常的新生儿如出生后显示淋巴样水肿,主动脉缩窄,小下鄂骨,也建议接受血染色体检查。2、儿童期:父母多注意身材矮小、生长缓慢或青春期延迟(14岁无性征发育或16岁无初潮),低发际,低位耳的女童,及早到医院诊治。经笔者统计,大多数儿童期就诊的患者的就诊原因是父母发现身材矮小或生长缓慢。Turner’s综合征的女孩如未经治疗,比正常人矮10-20cm;另外由于先天性卵巢功能衰竭,这些女孩缺乏第二性征,成年后还要面临不育的苦恼。因此,及早诊治以改善她们的身高和第二性征非常重要。有些女孩到了17、18岁因原发性闭经才来就诊,已错失了最好的治疗时间。笔者见过1个女孩,由于生长缓慢在4岁时被确诊,由于治疗开始得早,女孩20岁时身高达162 cm,已是一所名牌大学的学生了。一旦通过染色体检查确诊后,可以用生长激素促进身高直至达到理想身高或长速低于每年4cm,在达到青春发育启动年龄(13岁左右)后,可以性激素替代治疗,促进性征发育。性激素替代治疗方案必须在医生指导下谨慎进行,先用低剂量激素(经口服或皮下)治疗,1-2年后剂量增加,并加用孕激素行周期疗法。激素替代疗法不仅能维持第二性征和月经,而且可以预防低雌激素性疾病(如骨质疏松),可以持续到50岁,同时随访肝肾功能,血脂血凝情况,预防长期用药的副作用。虽然Turner’s综合征的患者平均智力正常,但有10%的女孩存在一定程度的心理发育延迟,有些女孩在视觉空间能力(视觉想象,直觉和地图理解方面),视觉注意力,问题解决能力方面存在困难。部分女孩在非语言交流方面能力差,这些现象在染色体为纯45x的患者中更常见。家长在教育孩子时,一定要多注重孩子在这些方面的训练。另外,这些孩子在青少年时期由于性发育幼稚导致自尊心受损和孤独,需要等多的心理咨询和关怀。当这些“小”女孩成年后,生育是她们面临的最大挑战,国外一项对276名Turner’s综合征的妇女的研究显示,87.7%终生无孩,其他的通过领养、自然受孕(仅占3%)或辅助生殖技术拥有孩子。国外学者已着手研究Turner’s综合征的生育能力保留:那些经B超能发现少量可见的,非条索化的卵巢的患者(基因核型多为嵌合型),在青春期有明显的卵巢生长和卵泡发育。但这些卵巢中仅有的残留卵泡凋亡速度很快,到青春期末,卵巢功能基本衰竭。因此专家主张,对这些患者,应在青春期冰冻卵子,成年后再融化复苏,受精后移植入宫腔。但在取卵前,这些患者必须补充性激素以达到一定的子宫发育程度,这些患者即使受孕后也面临着多于常人的考验:30%左右发生流产,20-25%发生胎儿畸形(含染色体畸形),22-50%发生胎儿宫内发育迟缓或早产。同时孕妇自己也面临着很多风险:40%左右发生妊娠糖尿病,妊娠期高血压疾病等,比较严重的并发症包括先天性心脏病恶化导致的心衰、动脉囊性中层坏死等。因此,Turner’s综合征的妇女准备怀孕前都要进行生育前检查,推荐内容如下:1、心脏核磁共振,心电图,心动图,肢体大动脉血压监测2、甲状腺功能检测,包括一些抗体指标3、空腹血糖,糖化血红蛋白;肝功能检测,如果异常还要进行肝脏B超。4、肾脏B超,如何存在肾脏畸形或高血压还要进行肾功能检测5、妇科盆腔B超如果通过辅助生殖的方式受孕(无论是通过自己还是代孕母亲),只要胚胎受孕自Turner’s综合征妇女自己的卵子,需行胚胎种植前的产前诊断,排除染色体异常的胚胎。在整个孕期,母儿都需要接受严密的监护。尤其是依靠自己卵子受孕的妇女,自然流产率和胎儿染色体异常的发生率高,必须接受包括羊水染色体检查在内的产前诊断。Turner’s综合征患者成年后还存在生育以外的医学问题,心血管疾病,自身免疫疾病和骨骼肌肉异常,视力缺损,听力丧失,口腔畸形更常见,要接受定期的健康体检。另外,这些妇女在恋爱婚姻方面的困难远比正常妇女大,心理问题仍不能忽视。有1名患病妇女曾很沮丧地说,她觉得周围人都把她当成未成熟的孩子,所有她不敢和男性深入交往。所以,这些妇女需要社会给予更多的关心。 德馨医院周日门诊,方便在校学生就诊,为青少年女性护航。医生介绍王哲蔚,德馨医院门诊时间:周日下午,预约电话18901703169
王哲蔚医生的科普号 2020年10月18日3711
0
1
2020年10月18日3711
0
1
-
《染色体病其实没什么可怕的!》徐志泉 海南省妇女儿童医学中心
《染色体病其实没什么可怕的!》————如果没防住!那只能坦然面对,然后充分重视!徐志泉 海南省妇女儿童医学中心 肾病风湿免疫科海南省妇女儿童医学中心儿童肾病风湿免疫科徐志泉正常人一共有23对染色体,父
徐志泉医生的科普号 2020年07月29日4001
0
8
2020年07月29日4001
0
8
特纳综合征相关科普号

李文京医生的科普号
李文京 主任医师
首都医科大学附属北京儿童医院
内分泌遗传代谢中心
115粉丝4.9万阅读

儿童健康快车~
梁丽俊 主任医师
宁夏医科大学总医院
儿科
1953粉丝14.7万阅读

张月萍医生的科普号
张月萍 主任医师
上海交通大学医学院附属澳门美高梅网站/仁济医院(北院)
生殖医学中心
5811粉丝28.9万阅读
-

 推荐热度5.0朱子阳 主任医师南京医科大学附属儿童医院 内分泌科
推荐热度5.0朱子阳 主任医师南京医科大学附属儿童医院 内分泌科矮小症 469票
性早熟 122票
发育迟缓 61票
擅长:矮小症、性早熟、糖尿病、甲状腺疾病、肾上腺疾病等小儿内分泌疾病的诊治。 -
 推荐热度4.3韩丽萍 主任医师郑大一附院 妇科
推荐热度4.3韩丽萍 主任医师郑大一附院 妇科子宫肌瘤 11票
妇科病 5票
卵巢囊肿 5票
擅长:擅长各类妇科微创手术及妇科肿瘤的规范化治疗;积极推广妇科单孔腹腔镜手术,在不孕症及子宫纵膈、宫腔粘连、子宫肌瘤、子宫脱垂等疾病方面积累了丰富的经验。对女性内分泌失调如不孕症、多囊卵巢综合征、闭经、功血、更年期综合征及生殖器官畸形等疾病有着扎实的诊治经验。 -
 推荐热度4.2潘洁雪 副主任医师复旦大学附属妇产科医院 妇科内分泌与生殖医学科
推荐热度4.2潘洁雪 副主任医师复旦大学附属妇产科医院 妇科内分泌与生殖医学科不孕不育 2票
特纳综合征 1票
擅长:不孕症,辅助生殖技术,多囊卵巢综合征,排卵障碍的诊治